 可咨詢
可咨詢
擅長:高危兒、神經系統病變、腦性癱瘓綜合征、抽動障礙、腦性癱瘓、抽動癥、缺氧缺血性腦病、顱腦損傷、精神發育遲緩、內分泌失調、性早熟、矮小癥、發育障礙、兒童多動癥、佝僂病、侏儒癥、多動癥、自閉癥、發育遲緩、智力低下、肺炎、支氣管炎、上呼吸道感染、感冒、功能性消化不良、消化不良、便秘、急性腸胃炎、新生兒黃疸、新生兒缺氧缺血性腦病
 可咨詢
可咨詢
擅長:呼吸系統病變、消化系統疾病、反復呼吸道感染、消化不良、感冒、過敏性鼻炎、支原體感染、腹瀉病、便秘、功能性消化不良、小兒腹瀉病、兒童多動癥、消化道紊亂、嬰兒腹瀉、胃炎、慢性腸炎、神經系統病變、抽動癥、腦性癱瘓、精神發育遲緩、小兒癲癇、性早熟、矮小癥、肺炎、腎病綜合征、哮喘、支氣管炎、自閉癥、濕疹、遺尿癥
 可咨詢
可咨詢
擅長:神經系統病變、偏癱、腦病、特發性面神經癱瘓、腦性癱瘓綜合征、面肌痙攣、頑固性失眠、脊柱、脊髓損傷、脊髓損傷、癡呆、腦外傷、周圍神經病
 可咨詢
可咨詢
擅長:呼吸系統病變、哮喘、感冒、肺炎、神經系統病變、小兒癲癇、癲癇、腦性癱瘓、抽動癥、熱性驚厥、精神發育遲緩、發育障礙、自閉癥、兒童多動癥、佝僂病、多動癥、發育遲緩、智力低下、消化系統疾病、便秘、食物過敏、嬰兒腹瀉、新生兒疾病、新生兒黃疸、新生兒窒息、感染性疾病、皰疹性咽峽炎、水痘、腮腺炎、內分泌失調、矮小癥、注意缺陷障礙
 可咨詢
可咨詢
擅長:鼻炎、上呼吸道感染、反復呼吸道感染、小兒肺炎、消化不良、小兒泄瀉、便秘、小兒腸炎、自閉癥、兒童多動癥、消化系統疾病、抽動障礙、呼吸系統病變、發育障礙、慢性咳嗽、神經系統病變、高危兒、化膿性腦炎、胃炎、周圍神經損傷、腦神經損傷、內分泌失調、發育遲緩、多動癥、顱內感染、精神發育遲緩、哮喘、新生兒缺氧缺血性腦病、腦性癱瘓、性早熟
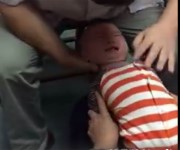
嬰幼兒頭部支撐弱,當被人用力搖晃時,頭部會不由自主地轉動。這種劇烈的晃動使孩子的大腦在顱骨內來回震蕩,造成腦部損傷,嚴重時可導致死亡。
2017-08-26 育兒




“鬼壓床”在醫學上稱為睡眠癱瘓癥(睡眠麻痹)。試著不要讓自己太累,不要熬夜,維持正常的作息,這樣通常就能減少睡眠麻痹癥的發生。不過,如果睡眠麻痹癥發生的頻率過高,影響睡眠質量,就應該找專業醫生來幫忙,看看是不是有其他問題,必要時采取藥物治療。
2017-05-24 生活Copyright ? 2011-2025 北京春雨天下軟件有限公司 All Rights Reserved
京ICP證120150號 京ICP備12050281號-1
(京)網藥械信息備字(2023)第 00530 號
違法和不良信息舉報:4000018855轉9 feedback@chunyu.me






